
A post by Cecina Babich Morrow, PhD student on the Compass programme.
Introduction
Sensitivity analysis seeks to understand how much changes in each input affect the output of a model. We want to be able to determine how variation in a model’s output can be attributed to variations in its input. Given the high amount of uncertainty present in most real-world modelling settings, it is crucial to understand the magnitude of this uncertainty’s impact on model results. Knowing how sensitive a model is to a particular parameter can help guide modellers in prioritising what level of precision is needed in estimating that parameter in order to produce valid results. Sensitivity analysis thus serves as a vital tool for modellers in numerous fields, allowing them to assess robustness and to identify key drivers of uncertainty. By systematically analysing the relative amount of influence that each input parameter has on the output, sensitivity analysis reveals which parameters have the greatest impact on the results.
By identifying these critical parameters, stakeholders can prioritize investments in data collection, parameter estimation, and uncertainty reduction. This targeted approach ensures that efforts are concentrated where they will have the most significant impact.
Why use Regional Sensitivity Analysis?
In this blog post, I will focus on one particular sensitivity analysis method that I have been using in my project so far to help understand the sensitivity of an output decision to input parameters which affect that decision. Regional Sensitivity Analysis (RSA) was developed in the field of hydrology, but has widespread applications in environmental modelling, disease modelling, and beyond.
My research focuses on environmental decision-making, so I frequently deal with models that output a decision that can take on one of several discrete values. For example, consider trying to make a decision about what to wear based on the weather. To make our decision, we use three input parameters about the weather: temperature, humidity, and wind speed. Then, our decision model can output one of three decisions: (1) stay home, (2) leave the house with a jacket, (3) leave the house without a jacket. We might then be interested in how sensitive our model is to each of our three weather-related input parameters to understand how much each one contributes to uncertainty in our ultimate decision. In this type of setting, we need to use a sensitivity analysis method that can handle continuous inputs, e.g. temperature, in conjunction with a discrete output, e.g. our decision.
For settings such as these where the inputs of our model are continuous and the outputs are discrete, RSA, also referred to as Monte Carlo filtering, is a potential method of sensitivity analysis [1]. RSA aims to identify which regions of input space corresponding to specific values in the output space [2, 3]. Originally, the method was developed in the field of hydrology for cases where the output variable is binary, or made such by applying a threshold. It has since been extended by splitting the parameter space into more than two groups [3, 4]. RSA is well-suited to sensitivity analysis in the case where the output variable is categorical [5].
RSA is fundamentally a Bayesian approach. First, prior distributions are assigned to the input parameters. The model is then run multiple times, sampling input parameters from these priors, and recording the resulting output values. By analysing the relationship between input uncertainties and output uncertainties, RSA identifies which parameters significantly affect the model’s predictions.
How does RSA work?
We will present the mathematical formalisation of RSA in a setting where we have a discrete output variable 


![\mathbf{x} = [x_1, x_2, \ldots, x_d]](https://s0.wp.com/latex.php?latex=%5Cmathbf%7Bx%7D+%3D+%5Bx_1%2C+x_2%2C+%5Cldots%2C+x_d%5D&bg=ffffff&fg=000000&s=0 "\mathbf{x} = [x_1, x_2, \ldots, x_d]")

Then, RSA compares the empirical conditional cumulative distribution functions (CDFs) 



")



 = F(x_i | y_1) = \ldots = F(x_i | y_m)")
The difference between these CDFs can be measured using several possible sensitivity indices. Typically, the Kolmogorov-Smirnov (KS) statistic is applied over all possible values of
![\text{stat}_{j,k} [KS(x_i)] = \text{stat}_{j,k} \left[\max_{x_i} \left \lvert F_{x_i | y_j} (x_i | y = y_j) - F_{x_i | y_k} (x_i | y = y_k) \right \rvert\right]](https://s0.wp.com/latex.php?latex=%5Ctext%7Bstat%7D_%7Bj%2Ck%7D+%5BKS%28x_i%29%5D+%3D+%5Ctext%7Bstat%7D_%7Bj%2Ck%7D+%5Cleft%5B%5Cmax_%7Bx_i%7D+%5Cleft+%5Clvert+F_%7Bx_i+%7C+y_j%7D+%28x_i+%7C+y+%3D+y_j%29+-+F_%7Bx_i+%7C+y_k%7D+%28x_i+%7C+y+%3D+y_k%29+%5Cright+%5Crvert%5Cright%5D+&bg=ffffff&fg=000000&s=0 "\text{stat}_{j,k} [KS(x_i)] = \text{stat}_{j,k} \left[\max_{x_i} \left \lvert F_{x_i | y_j} (x_i | y = y_j) - F_{x_i | y_k} (x_i | y = y_k) \right \rvert\right]")
where 

For instance, consider the following situation with an input parameter ")
")
")
")


Figure 1. Visualisation of RSA using a summary statistic of the KS statistic as a sensitivity index. The blue, green, and red distributions are the empirical conditional CDFs ")

As an alternative to using the KS statistic, we can instead apply a statistic to spread, i.e. the area between the CDFs:
![\text{stat}_{j,k} [\text{spread}(x_i)] = \text{stat}_{j,k} \left[ \int_{-\infty}^\infty \max \left(F_{x_i | y_j} (x_i | y = y_j), F_{x_i | y_k} (x_i | y = y_k)\right) dx_i - \int_{-\infty}^\infty \min \left(F_{x_i | y_j} (x_i | y = y_j), F_{x_i | y_k} (x_i | y = y_k)\right) dx_i \right]](https://s0.wp.com/latex.php?latex=%5Ctext%7Bstat%7D_%7Bj%2Ck%7D+%5B%5Ctext%7Bspread%7D%28x_i%29%5D+%3D+%5Ctext%7Bstat%7D_%7Bj%2Ck%7D+%5Cleft%5B+%5Cint_%7B-%5Cinfty%7D%5E%5Cinfty+%5Cmax+%5Cleft%28F_%7Bx_i+%7C+y_j%7D+%28x_i+%7C+y+%3D+y_j%29%2C+F_%7Bx_i+%7C+y_k%7D+%28x_i+%7C+y+%3D+y_k%29%5Cright%29+dx_i+-+%5Cint_%7B-%5Cinfty%7D%5E%5Cinfty+%5Cmin+%5Cleft%28F_%7Bx_i+%7C+y_j%7D+%28x_i+%7C+y+%3D+y_j%29%2C+F_%7Bx_i+%7C+y_k%7D+%28x_i+%7C+y+%3D+y_k%29%5Cright%29+dx_i+%5Cright%5D&bg=ffffff&fg=000000&s=0 "\text{stat}_{j,k} [\text{spread}(x_i)] = \text{stat}_{j,k} \left[ \int_{-\infty}^\infty \max \left(F_{x_i | y_j} (x_i | y = y_j), F_{x_i | y_k} (x_i | y = y_k)\right) dx_i - \int_{-\infty}^\infty \min \left(F_{x_i | y_j} (x_i | y = y_j), F_{x_i | y_k} (x_i | y = y_k)\right) dx_i \right]")
where 
Higher values of either sensitivity index for a given input parameter 
")
")

Figure 2. Comparison of sensitivity of a model to two input parameters, ")
Extensions of RSA
One notable limitation of RSA, identified since its inception [2], is its inability to handle parameter interactions. A zero value of the sensitivity index is a necessary condition for insensitivity, but it is not sufficient [2, 5]. Inputs that contribute to variation in the model output only through interactions can have the same univariate conditional CDFs, and thus RSA cannot properly identify their impact on model output. For theoretical examples, see Fig. 1 of [2] and Example 6 of Section 5.2.3 in [1]. In our toy example, we may have a decision model where the output decision is not particularly sensitive to temperature or humidity on their own, but it may be very sensitive to an interaction between these two parameters since their combined effects impact how warm or cool the weather actually feels.
In situations such as these where interactions between input variables may matter more than each variable on its own, RSA can be useful for ranking, but it cannot be used for screening, another goal of sensitivity analysis aiming to identify variables with little to no influence on output variability[1, 5]. To address this limitation, RSA can be augmented with machine learning methods such as random forests and density estimation trees [6]. Spear et al. performed a sensitivity analysis of a dengue epidemic model to demonstrate how these tree-based models can augment RSA [6].
First, the authors performed RSA in its original form, using the KS statistic to examine the difference between the univariate CDFs. Then, they used random forest analysis to classify model runs into the various output values. Then, a measure of variable importance, such as Gini impurity, was used to rank the input parameters in terms of their influence on the model output [6]. Random forest allows for the incorporation of the effects of variable interactions in ranking the importance of each parameter. By comparing the parameter ranking resulting from RSA with that from the random forest, they identified parameters which impacted the output through interaction. Finally, they used density estimation trees to help identify regions of parameter space corresponding to particular output values. Density estimation trees are the analogue of classification and regression trees, instead attempting to estimate the probability density function that gave rise to a particular region of output space [7]. By applying density estimation trees as part of the sensitivity analysis, Spear et al. were able to examine the effects of scale on sensitivity, identifying parameters which may be relatively unimportant when ranking across the entire parameter subspace, but are highly influential in small subspaces.
Further research such as this highlights the benefits of combining multiple sensitivity analysis methods in order to gain a full picture of how model inputs affect uncertainty in the output.
Conclusions
Hopefully this blog has been an informative crash course in regional sensitivity analysis! Note that the visualisations in this post have been created using the SAFEpython toolbox [8]. If you have any questions or comments, please feel free to get in touch at cecina.babichmorrow@bristol.ac.uk.
References
[1] A. Saltelli, Global sensitivity analysis: the primer. Wiley, 2008. [Online]. Available: https://onlinelibrary.wiley.com/doi/book/10.1002/9780470725184
[2] R. Spear and G. Hornberger, “Eutrophication in peel inlet—II. identification of critical uncertainties via generalized sensitivity analysis,” Water Research, vol. 14, no. 1, pp. 43–49, 1980. [Online]. Available: https://www.sciencedirect.com/science/article/pii/0043135480900408
[3] J. Freer, K. Beven, and B. Ambroise, “Bayesian estimation of uncertainty in runoff prediction and the value of data: An application of the GLUE approach,” Water Resources Research, vol. 32, no. 7, pp. 2161–2173, 1996. [Online]. Available: https://onlinelibrary.wiley.com/doi/abs/10.1029/95WR03723
[4] T. Wagener, D. P. Boyle, M. J. Lees, H. S. Wheater, H. V. Gupta, and S. Sorooshian, “A framework for development and application of hydrological models,” Hydrology and Earth System Sciences, vol. 5, no. 1, pp. 13–26, 2001. [Online]. Available: https://hess.copernicus.org/articles/5/13/2001/
[5] F. Pianosi, K. Beven, J. Freer, J. W. Hall, J. Rougier, D. B. Stephenson, and T. Wagener, “Sensitivity analysis of environmental models: A systematic review with practical workflow,” Environmental Modelling & Software, vol. 79, pp. 214–232, 2016. [Online]. Available: https://linkinghub.elsevier.com/retrieve/pii/S1364815216300287
[6] R. C. Spear, Q. Cheng, and S. L. Wu, “An example of augmenting regional sensitivity analysis using machine learning software,” vol. 56, no. 4, p. e2019WR026379. [Online]. Available: https://onlinelibrary.wiley.com/doi/abs/10.1029/2019WR026379
[7] P. Ram and A. G. Gray, “Density estimation trees,” in Proceedings of the 17th ACM SIGKDD international conference on Knowledge discovery and data mining. ACM, pp. 627–635. [Online]. Available: https://dl.acm.org/doi/10.1145/2020408.2020507
[8] F. Pianosi, F. Sarrazin, and T. Wagener, “A Matlab toolbox for global sensitivity analysis,” Environmental Modelling & Software, vol. 70, pp. 80–85, 2015. [Online]. Available: https://linkinghub.elsevier.com/retrieve/pii/S1364815215001188


 with a particular sequence, we can take a sample from this synthesis and analyse it via mass spectrometry. In this process, molecules in the sample are first fragmented — broken apart into ions — and these charged fragments are then passed through an electromagnetic field. The trajectory of each fragment through this field depends on its mass/charge ratio (m/z), so measuring these trajectories (e.g. by measuring time of flight before hitting some detector) allows us to calculate the m/z of fragments in the sample. This gives us a discrete mass spectrum: counts of detected fragments (intensity) across a range of m/z bins [5].
with a particular sequence, we can take a sample from this synthesis and analyse it via mass spectrometry. In this process, molecules in the sample are first fragmented — broken apart into ions — and these charged fragments are then passed through an electromagnetic field. The trajectory of each fragment through this field depends on its mass/charge ratio (m/z), so measuring these trajectories (e.g. by measuring time of flight before hitting some detector) allows us to calculate the m/z of fragments in the sample. This gives us a discrete mass spectrum: counts of detected fragments (intensity) across a range of m/z bins [5].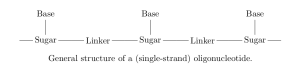
 every product of a single fragmentation of
every product of a single fragmentation of 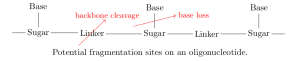
 , where
, where  is the intensity in the
is the intensity in the  bin. For a set
bin. For a set  of possible fragments, let
of possible fragments, let  that is actually present. We would like to estimate the amounts of each fragment based on the spectrum
that is actually present. We would like to estimate the amounts of each fragment based on the spectrum  .
. and
and  and this produced a spectrum
and this produced a spectrum  , we can say the intensity contributed to bin
, we can say the intensity contributed to bin  In mass spectrometry, the intensity in a single bin due to a single fragment is linear in the amount of that fragment, and the intensities in a single bin due to different fragments are additive, so in some general spectrum we have
In mass spectrometry, the intensity in a single bin due to a single fragment is linear in the amount of that fragment, and the intensities in a single bin due to different fragments are additive, so in some general spectrum we have 
 such that
such that  (so the columns of
(so the columns of  correspond to fragments in
correspond to fragments in  solves
solves  . In practice this exact solution is not found — due to experimental noise and potentially because there are contaminant fragments in the sample not included in
. In practice this exact solution is not found — due to experimental noise and potentially because there are contaminant fragments in the sample not included in  for which
for which  is close to
is close to  -normalise these columns, meaning the total intensity (over all bins) of each fragment in the library matrix is uniform, and so the values in
-normalise these columns, meaning the total intensity (over all bins) of each fragment in the library matrix is uniform, and so the values in  can be directly interpreted as relative abundances of each fragment.
can be directly interpreted as relative abundances of each fragment.}=\left(\mathbf A^T \frac{\mathbf b}{\mathbf{A\hat x}^{(t)}}\right)\odot \mathbf{\hat x}^{(t)}.")
 represent (respectively) elementwise division and multiplication of two vectors. This is known as the Richardson-Lucy algorithm [9].
represent (respectively) elementwise division and multiplication of two vectors. This is known as the Richardson-Lucy algorithm [9].}=\left(\mathbf A^T \frac{\mathbf b}{\mathbf{A\hat x}^{(t)}}\right)\odot \frac{ \mathbf{\hat x}^{(t)}}{\mathbf 1 + \lambda},")
 is a regularisation parameter [10].
is a regularisation parameter [10]. , we smooth and bin the m/z values of the most abundant isotopes of
, we smooth and bin the m/z values of the most abundant isotopes of  , and store these values in the columns of
, and store these values in the columns of  ) for clarity.
) for clarity.


